

1.致癌蛋白ALK通过磷酸化SMAD4致TGF-β信号失活的研究
(Nat Cell Biol. 2019 Feb;21(2):179-189. )
TGF-b信号通路在癌症发生的早期具有重要抑癌

作用,但癌细胞自身常会建立起许多逃逸TGF-b信号的分子机制。SMAD家族蛋白是TGF-b信号通路的中心蛋白,其中核心蛋白SMAD4在胰腺癌和一些消化道肿瘤中会发生高频率的缺失和突变,然而在很多其它类型的癌症中却极少出现SMAD4基因的变异,这表明肿瘤细胞中可能存在其它导致TGF-b信号失活的调控机制。
本课题组在前期的研究工作中筛选到了可以直接磷酸化修饰SMAD4并使其失活的酪氨酸激酶,即间变性大细胞淋巴瘤激酶ALK(Anaplastic Lymphoma Kinase)。 ALK是一个原癌基因,它可以通过突变或染色体异位的方式产生出具有组成性激酶活性的各种变异体。ALK的变异体可以在很多不同类型的癌症肿瘤中表达,进而驱动癌症的发生发展。最常见的ALK阳性肿瘤有间变性淋巴癌、非小细胞肺癌、神经母细胞瘤和炎性乳腺癌等等。
深入的研究发现ALK

阳性肿瘤中SMAD4存在高度磷酸化的现象,同时证实了该磷酸化是由ALK在SMAD4的Tyr95位点直接修饰形成的。ALK磷酸化SMAD4并抑制TGF-b信号的这一调控效应不仅仅存在于表达NPM-ALK的间变性大细胞淋巴瘤中;在表达EML4-ALK的非小细胞肺癌及表达ALK-F1174L的神经母细胞瘤中也同样存在,即ALK对TGF-b的调控具有广泛性。
在分子调控机制上,我

们发现ALK对SMAD4的磷酸化不影响SMADs复合物的形成、入核;也不影响SMADs与转录共激活因子p300/CBP的相互作用;但是会直接影响SMAD4与DNA的结合能力,从而抑制TGF-b信号下游肿瘤抑制基因的表达,导致TGF-b通路的肿瘤抑制功能丧失。通过ALK抑制剂或利用遗传干扰技术降低ALK表达均能降低SMAD4的此类磷酸化,恢复ALK阳性肿瘤细胞对TGF-b
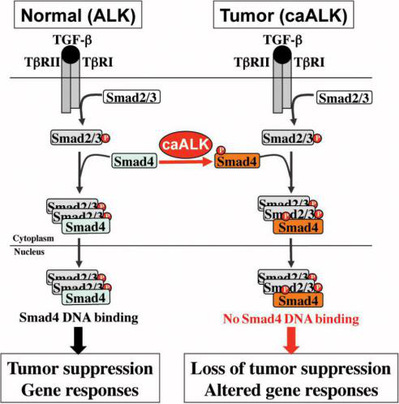
信号通路的响应。最后,在人类淋巴瘤样本中还检测发现了SMAD4的Tyr95位点磷酸化的水平与ALK的表达量呈正相关变化。揭示了在肿瘤发生过程中存在SMAD4酪氨酸激酶磷酸化,这为TGF-b信号在肿瘤中的失活提供了一种新的机制,并为ALK阳性癌症的靶向治疗提供了新指导。
2.Smad7通过促进STAT3信号维持小鼠胚胎干细胞的全能性
(Proc Natl Acad Sci U S A. 2017 Sep 19;114(38):10113-10118.)
TGF-b超家族在调控细胞潜能及分化过程中发挥了重要作用。众所周知,Smad7作为TGF-b信号通路的负反馈产物,精密调控着TGF-b诱导的一系列生物学反应。但是,Smad7不依赖于TGF-b信号通路的非经典功能却鲜有报道。

本课题组发现Smad7在未分化的小鼠ESC中高表达,随着细胞的分化,其表达水平逐渐下降。在拟胚体分化实验中,过表达Smad7能够有效遏制干性基因的下降,而敲减Smad7则会显著抑制小鼠ESC的自我更新,并且促进ESC向外胚层和中胚层分化。在重编程的过程中,伴随着全能性基因的表达,Smad7的表达水平也会上升。如果敲减掉Smad7会明显减少iPSC克隆的形成。因此,Smad7不仅能够促进小鼠胚胎干细胞的自我更新,而且对可诱导多能干细胞的重编程过程也有重要影响。
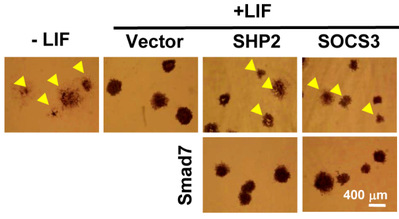
有意思的是,我们发现Smad7对ESC全能性的调控并不依赖于它对TGF-b/BMP的抑制作用。利用质谱方法筛选Smad7在小鼠ESC中潜在的相互作用蛋白,并通过免疫共沉淀(Co-IP)进行验证,确认 Smad7与IL-6家族成员的共受体gp130存在直接相互作用。并且,Smad7在gp130上的结合区域恰好与gp130-STAT3的负调控因子SHP2/SOCS3的结合位点(Y759)重合。进一步的体外体内实验也都表明Smad7

确实能够抑制SHP2/SOCS3与gp130的结合。最后还通过一系列实验证明了Smad7通过拮抗SHP2/SOCS3对LIF-STAT3信号通路的抑制作用,从而增强STAT3信号,维持ESC的自我更新。该研究揭示了Smad7与STAT3信号的相互作用对于维持小鼠胚胎干细胞全能性的关键作用,同时也提示这一分子机制极有可能在其他生理病理过程,如炎症反应以及肿瘤中有更为广泛的影响。
3.PTPN3通过稳定TbRI促进TGF-b信号通路
(EMBO J, 2019)
转化生长因子b (Transforming Growth Factor-b,TGF-b) 在细胞增殖、分化、凋亡以及胚胎发育等一系列生理过程中发挥着重要的调控作用。TGF-b
 信号通路的异常调控可能引发多种疾病,如免疫性疾病、心血管疾病、胚胎发育异常和肿瘤等,因而研究TGF-b信号通路的精准调控对于癌症等疾病治疗具有深远意义。
信号通路的异常调控可能引发多种疾病,如免疫性疾病、心血管疾病、胚胎发育异常和肿瘤等,因而研究TGF-b信号通路的精准调控对于癌症等疾病治疗具有深远意义。
本课题组在前期筛选中发现了一个新的TGF-b信号通路的重要调控因子,非受体型蛋白质酪氨酸磷酸酶3 (Protein Tyrosine Phosphotase 3, PTPN3)。过表达PTPN3能显著增强TGF-b诱导的R-Smad磷酸化和下游基因响应。进一步的机制研究表明, PTPN3通过其FERM结构域抑制TGF-b I型受体(TbRI)与E3泛素连接酶Smurf2的相互作用,从而降低TbRI的泛素化水平,阻止TbRI经蛋白酶体途径发生降解,增加了TbRI的蛋白稳定性。并且,PTPN3的磷酸酶失活突变
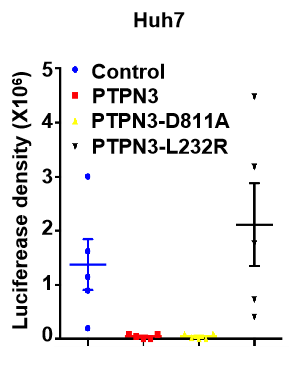 体D811A (asparticacid-to-alanine substitution at amino acid residue 811) 和野生型一样,能够增强TGF-b信号通路在细胞水平的响应及其对肿瘤形成的抑制
体D811A (asparticacid-to-alanine substitution at amino acid residue 811) 和野生型一样,能够增强TGF-b信号通路在细胞水平的响应及其对肿瘤形成的抑制
用,这表明PTPN3对TGF-b信号通路的调控作用并不依赖于其磷酸酶活性。有趣的是,我们发现PTPN3的另一个突变体L232R (leucine-to-arginine substitution at amino acid residue 232)可以使PTPN3对TGF-b信号通路的促进作用完全丧失。L232R恰好位于PTPN3发挥功能的FERM结构域,并且它是一个肝内胆管癌中的高频突变体。我们推测极有可能是L232关键位点的突变,导致PTPN3丧失了对TGF-b抑制肿瘤细胞生长的正向调控作用,从而促进肝内胆管癌的发生发展。此外,生物信息学分析结果也表明,在肝癌病人的肿瘤样本中PTPN3的mRNA水平和正常样本相比显著下降,并且PTPN3表达水平低的肝癌病人的生存率更低。以上结果均表明,PTPN3可能在肝癌中作为一个肿瘤抑制因子,通过调控TGF-b信号通路发挥作用。本研究发现了新的TGF-b信号通路调控因子PTPN3,
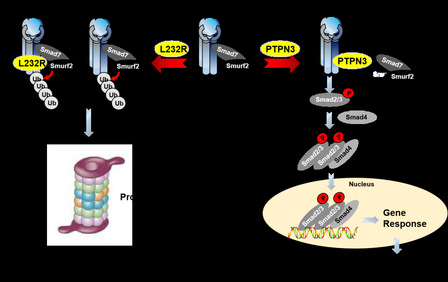
证实PTPN3在生理和病理情况下对TGF-b信号通路具有重要的调控作用,并拓展了PTPN3磷酸酶之外的新功能,为找到潜在的肝癌防治手段,提供了新的理论基础。